
了解蛋白质的三级结构对于理解其生物学功能和发现新型疗法至关重要。蛋白质结构的计算预测是X射线晶体学、核磁共振和低温电子显微镜等实验方法的有力替代和补充方法。蛋白质结构预测的一项重要任务是识别具有相似三级结构的蛋白质,这些蛋白质可用作模板来模拟另一个蛋白质的未知结构。识别这些结构相似的蛋白质的过程称为折叠识别(或穿线),这是一种预测查询蛋白质结构的有效方法,尤其当查询蛋白质与其他已知结构的蛋白质在序列水平上具有较低的同一性(即<25%)时。
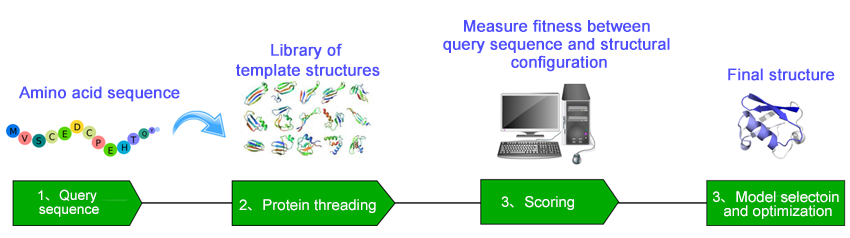
迈博睿生物凭借其专业知识,通过将序列谱-谱比对与多种结构信息相结合,帮助识别目标蛋白在已知模板蛋白质结构中的正确结构折叠。我们的方法首先构建结构模板数据库/库,然后逐步用未知结构的查询序列替换库中已知蛋白质结构的序列。通过优化设计的评分函数,将目标序列与每个结构模板进行比对。针对模板数据库中所有已知的三维结构重复此过程,直到找到最佳拟合。选择统计上最可能的比对作为线程预测,并通过将目标序列的骨架原子放置在所选结构模板的对齐骨架位置来构建目标结构模型。与仅进行序列比对不同,这些方法利用了三维结构信息提供的额外信息。生成的结构模型均经过质量验证,可用于下游计算机模拟或实验工作,例如蛋白质工程和药物设计。
通过折叠识别(线程)预测蛋白质结构
特征
用于远程同源性识别的精确序列和结构比对算法用于相似性比较的 1D-3D 轮廓
模板数据库构建
使用功能域信息和序列进化信息
基于知识的评分函数包含突变潜力、环境适应潜力、成对潜力、二级结构兼容性、间隙惩罚等。
多线程方法识别收敛解
将线程与其他结构预测方法相结合的混合方法
通过能量计算验证结构/序列匹配
输出结果包含模板蛋白信息、查询模板比对结果以及最重要的目标蛋白全长模型。我们还可以根据客户的具体需求定制服务,并将我们的计算程序集成到您的工作流程中。如需了解更多关于折叠识别服务的详细信息,请随时联系我们。



